
GxP Lifeline
Top Regulatory Trends Affecting Med Dev in Asia-Pacific Moving into 2019

Rapid regulatory change is normal in the emerging (and established) medical device markets of the Asia-Pacific. Substantial changes in 2018 were no exception, including in some cases, whole new regulatory systems rolled out across the region. And 2019 will be offering up more of the “different.”
The pattern is of increasing regulatory sophistication and controls. But it’s by no means smooth sailing. Some jurisdictions are becoming increasingly complex (e.g., China); some are in the very early stages of quite substantial change (e.g., India); and there are still a number of challenging countries that remain mostly unregulated.
A company’s decision to enter these economies should be treated with caution and ensure that the commercial opportunity justifies the risk. The mega-market of China is very tempting. But nothing is quite what it seems in China, where success demands a long-term commitment of pressure and downright hard work to navigate the intricacies of Chinese regulations. At the other end of the scale, lack of regulation is rarely a good thing for industry, as it usually goes hand in hand with lack of transparency and all sorts of commercial risks and uncertainty.
The beginning of 2019 saw substantial change with the introduction of new Indian regulations. It was a rocky start, made all the more complex by an obscure bureaucratic requirement for rotation of roles in the Indian public service that resulted in the replacement of key players just before the fire of the starting gun. All of that was hastily fixed and things settled down shortly thereafter with international manufacturers starting to gain traction on entry to the very large Indian market.
India offers two pathways for medical device approval, one for local manufacturers and a second for importers. The regulations are modelled on the Global Harmonisation Task Force (GHTF) and include a risk-based system of classification and regulatory review which all starts with a submission through an online system. Importers will be required to have local representation for their application and continued license holding.
We also saw continued changes in China culminating with a complete reorganization of the relevant ministries and the Chinese version of the U.S. Food and Drug Administration (FDA) being transformed into the country’s new National Medical Products Administration (NMPA). The changes were rapid fire and too numerous to list here, but highlights included extensions to clinical trial exemptions for many more devices and IVDs; commencement of an international audit program for foreign manufacturers; and changes to type testing requirements. China has recognized the crushing weight of the broad-based default of requiring local clinical trials and is rapidly winding back that expectation. It has floated a possible blanket exemption for all Class II devices in 2019. Other changes include the streamlining of ethics processes with adoption of a “lead site” process where all secondary sites are required to recognise the lead site’s review and approval of protocols.
The biggest challenge of 2018 in China for manufacturers was a complete logjam in type testing in a government operated laboratory as a mandatory prerequisite to filing for registration. However, a cancellation of fees by the country’s national industry ministry – ostensibly to provide benefits to manufacturers — was not managed well with no substitute funding provided to laboratories operated by provincial governments. This resulted in an effective cessation of testing for much of the year pending sorting out the funding. That’s all now been resolved, and labs are working through the backlogs, but it’s still painfully slow and astute applicants will shop around some of the lesser known labs outside main centers for faster service.
Most markets require a legal representative in the market to take responsibility for regulatory communications and compliance. That’s certainly true in China. However, NMPA is planning to step up the expectations on authorized agents, following the lead of Europe in requiring representatives to assume shared product liability with the manufacturer. Given China’s very stringent criminal justice system, that’s a major risk and is expected to test the commitment of regulatory agents to China.
In Australia, the Therapeutic Goods Administration (TGA) took a bold step in substantially broadening recognition of international approvals and embracing the Medical Device Single Audit Program (MDSAP). Up until now, Australia has for most devices and some IVDs accepted a CE marking certificate as the basis for Australian regulatory approval in place of a direct TGA assessment. TGA still does a desktop audit of key clinical evidence for higher risk devices and for the very highest risk devices containing medicines or biologicals, and conducts its own full conformity assessment. But in August 2018 all of that changed. TGA announced it would accept FDA, Canadian and Japanese approvals as suitable evidence for grant of inclusion on the Australian Register of Therapeutic Goods (ARTG). There were some caveats, the most important of which was that the manufacturer must hold either an MDSAP certificate or a Japanese quality systems certificate; a U.S. manufacturer registration and quality systems regulation (QSR) audit do not count.
TGA’s unilateral acceptance of four of the largest and most sophisticated international jurisdictions was a transformational step, which considerably advanced the cause of international harmonization. It also provided a big boost to the value of the MDSAP program, which is growing exponentially. These changes are particularly important in the context of the current challenges in Europe where the transition to the European Union’s Medical Device Regulation (MDR) has been anything but smooth. CE marks have been somewhat of a global regulatory passport. They are accepted as the basis of abridged assessments in many Asian jurisdictions (e.g., Taiwan, Singapore and Malaysia). However, CE “passports” are going to be much harder to get under the MDR. So, TGA’s wider ranging acceptance is very welcome news, which provides much greater flexibility for manufacturers to enter the Australian market and will be keenly watched by other jurisdictions.
There are some subtleties to the TGA acceptances (see graphic). Classifications must align; for example a Class III device in Australia can only be supported by a Premarket Approval Application (PMA) from the U.S., not a 510(k).
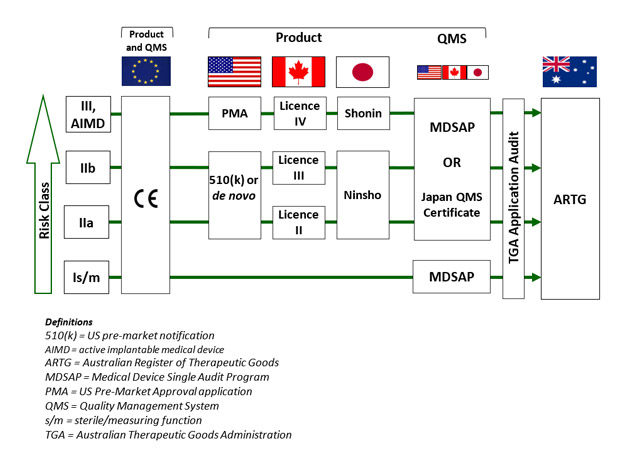
With the introduction of the MDSAP, there has been a growing interest from companies looking at this as a way of leveraging their international approvals. We have observed a substantial increase in the certification to MDSAP, which supports entry into several economies in Asia-Pacific. MDSAP is intended to provide a trusted audit which not only reviews manufacturing quality systems but also verifies the adequacy of system processes to ensure regulatory compliance. MDSAP is not as challenging as first feared and as the development of the program has proceeded, it has become more refined. Think of it as ISO 13485 with local flavors and nuances. A manufacturer with robust ISO 13485 can tackle MDSAP with incremental changes to systems.

Grant Bennett is CEO of Brandwood Biomedical and provides expert consultancy services relating to regulatory compliance in Australia, New Zealand and the Asia-Pacific. Grant is also responsible for management oversight of all Australian consulting projects.