
GxP Lifeline
What Verification and Validation Activities are Required for a First in Human Study?
Manufacturers preparing for a first in human (FIH) study of a new medical device face questions like these:
- Do we need to perform electrical safety testing of our new medical device prior to our upcoming first in human (FIH) study?
- What about biocompatibility or sterilization validation?
- What level of verification and validation (V&V) activities are required? What level of proof is needed to demonstrate that the risk mitigations are effectively implemented and the identified risks controlled for a device being used as part of a FIH study?
For an investigational device, manufacturers are exempt from the U.S. Food and Drug Administration’s (FDA) Quality System Regulations, except for the requirements for design control. Design V&V are key elements of design control that ensure the device meets its requirements and user needs. However, investigational devices are often neither full featured nor in their final format. It can therefore be difficult to determine which V&V activities are required for an investigational device and which can be deferred and performed on later iterations of the design.
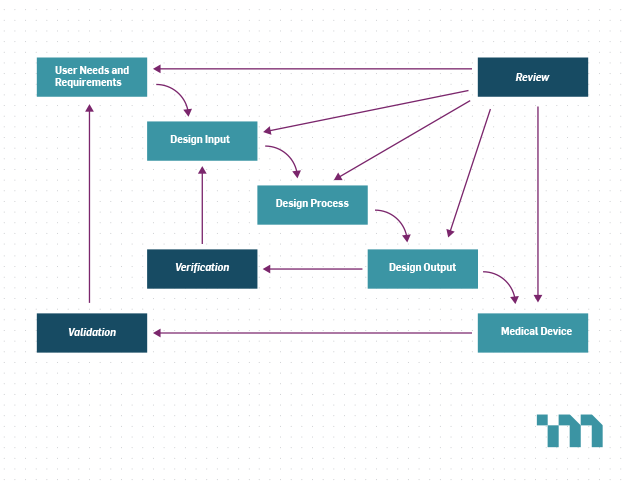
Prior to performing a clinical study of a medical device involving human subjects, the sponsor of the clinical study, often the device manufacturer, is responsible for obtaining Institutional Review Board (IRB) approval and Investigational Device Exemption (IDE) approval from the FDA for devices that pose a significant risk. Both the IRB and the FDA will evaluate the risks and benefits of the device to determine if the study is warranted. In other words, they are tasked with ensuring that due care has been used to minimize risks and maximize the likelihood of benefits.
Like so many things associated with medical devices, the best guidance in determining the appropriate V&V activities for an investigational device is to apply a risk-based approach. This approach should consider not only the risks to the user and patient, but also the risks to the study outcome.
Safety Risks to User/Patient
Investigational devices have a different risk profile than the envisioned commercial device. They are often neither full featured nor in their final format. They may incorporate off-the-shelf components rather than custom hardware and rely on lower volume manufacturing methods. Additionally, FIH studies are generally small in size with carefully selected patient populations. The study protocol can implement training and human-based risk control measures that may not be practical with a commercial device.
The first step in determining the appropriate V&V activities for an investigational device is to use the risk management process to fully identify the safety risks to the patient and users associated with the use of the device in the study and the mitigations for these risks. The V&V activities should then focus on demonstrating the device design meets the requirements related to these device risks and their mitigations. Examples of activities include:
- Bench testing of device function in simulated use and off-nominal conditions including verification of expected alerts and alarms.
- Electrical safety testing such as leakage current and dielectric strength.
- Characterization of the device’s electromagnetic emissions to demonstrate that the device won’t interfere with other equipment.
- Usability testing to demonstrate that any serious use-related risks have been mitigated.
- Validation of cleaning or sterilization methods, when patient contact with the device may lead to infection or other adverse reactions.
- Validation of the integrity of the packaging sterile boundary if devices are to be supplied sterile to the study site.
Additionally, biocompatibility testing should be considered in accordance with the risks associated with the duration and nature of patient contact. For some devices, it may be appropriate to mitigate these risks through the selection of materials known to have a low risk of biocompatibility issues. If prior use or testing has demonstrated the biocompatibility of a material, then the V&V activities may focus on reviewing the available data and performing chemical characterization to demonstrate that the selected material and manufacturing methods result in risks similar to those of the documented material.
Risks to the Study Outcome
Manufacturers should also consider V&V activities that demonstrate the device design is free from deficiencies that would interfere with the study outcome. Conditions that render the device non-functional or could lead to loss of data may present a low risk to the patient but can have a large impact on the study results because investigational devices often have additional data collection functions in support of the study. Examples of activities include:
- Testing of electromagnetic immunity to demonstrate that device function won’t be affected by the electromagnetic environment at the study site.
- Characterization of device function over the range of expected operating conditions (e.g., temperature, humidity and altitude).
Significant design changes after the study could render the results inapplicable to the commercial device. V&V activities performed early could identify and prevent major design changes later in the development lifecycle.
Final Step – Review the V&V Plan with the Institutional Review Board(s)
Institutional Review Boards (IRBs) will evaluate the risks and benefits of each investigational device submitted for their review, however each IRB may have different expectations regarding the level of proof required. This makes it difficult for manufacturers to anticipate what will be necessary to comply with these expectations. Therefore, drafting the V&V plan and reviewing it with the IRB before implementation is highly recommended. Applying a risk-based approach in determining the proposed V&V activities and clearly mapping the activities to device risks will facilitate these discussions. Ultimately, risk-based planning of V&V activities can help ensure a safe and informative FIH study.

Lynessa Erler is the director of testing and test systems in MPR Associates’s Health and Life Sciences group. She hold a bachelor’s degree in mechancial engineering form Virginia Tech. Since joining MPR in 1994, she has become a subject matter expert and provides technical leadership in verification and validation of medical and diagnostic device design and software development. Ms. Erler is also MPR’s senior national Instruments/LabVIEW expert and has applied this expertise in the development of product and test systems.